
Structure–property relationships of water adsorption in metal–organic frameworks†
Abstract
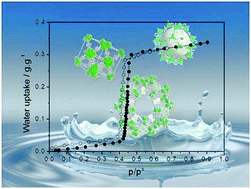
A set of 15 metal–organic frameworks (MIL-53, MIL-68, MIL-125, UiO-66, ZIF) exhibiting different pore size, morphology, and surface chemistry is used to unravel the numerous behaviors of water adsorption at room temperature in this class of materials. Outstanding “S”-shaped (type V) adsorption isotherms are observed for MIL-68 type solids. We show that the underlying mechanism of water adsorption can be rationalized using a simple set of three parameters: the Henry constant (i.e. the slope of the adsorption pressure in the low pressure range), the pressure at which pore filling occurs, and the maximum water adsorption capacity. While the Henry constant and pore filling pressure mostly depend on the affinity of water for the surface chemistry and on pore size, respectively, these two parameters are correlated as they both reflect different aspects of the hydrophobicity–hydrophilicity of the material. For a given type of porous structure, the functionalization of the material by hydrophilic moieties such as hydrogen bonding groups (amine or aldehyde) systematically leads to an increase in the Henry constant concomitantly with a decrease in the pore filling pressure. As for the adsorption mechanism, we show that, for a given temperature, there is a critical diameter (Dc ∼ 20 Å for water at room temperature) above which pore filling occurs through irreversible capillary condensation accompanied by capillary hysteresis loops. Below this critical diameter, pore filling is continuous and reversible unless the material exhibits some adsorption-induced flexibility.
 Please wait while we load your content...
Please wait while we load your content...

