
Understanding the adsorption behavior of surface active molecules on ZnO nanostructures by experimental and first-principles calculations
Abstract
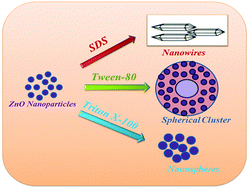
Zinc oxide (ZnO) nanostructures with different morphologies are prepared in the presence of surface active molecules such as sodium dodecyl sulphate (SDS), Tween 80 and Triton X-100 by a chemical method. The experimental and first principles methods are employed to understand the microscopic origin of the asymmetric growth mechanism of ZnO in the presence of various surface active molecules. Effect of increase in the amount of surface active molecules and temperature is studied on the growth morphology of ZnO. An innovative method is developed to synthesize ZnO nanowires (NWs) in the presence of SDS. Spherical nanoparticles (NPs) to spherical clusters are obtained in the presence of Triton X-100 and Tween 80. These results are then supported by first principles calculations. The adsorption of the –OH functional group on both polar and nonpolar surfaces of ZnO is modelled by using density functional theory (DFT). The calculated binding energy (BE) is almost equivalent on both the surfaces with no preference on any particular surface. The calculated value of BE shows that the –OH group is physio-adsorbed on both the surfaces. This results in the spherical morphology of nanoparticles prepared in the presence of Tween 80. Bader charge analysis shows that the charge transfer mainly takes place on top two layers of the ZnO(10![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0) surface. The absence of high values of electron localization function (ELF) reflects the lack of covalent bonding between the –OH group and the ZnO(100) surface.
0) surface. The absence of high values of electron localization function (ELF) reflects the lack of covalent bonding between the –OH group and the ZnO(100) surface.
 Please wait while we load your content...
Please wait while we load your content...

