
Recent advances in the design of antimicrobial peptide conjugates
 ac
and
Carlota O.
Rangel-Yagui
*a
ac
and
Carlota O.
Rangel-Yagui
*a
Abstract
Antimicrobial peptides (AMPs) are ubiquitous host defense peptides characterized by their antibiotic activity and lower propensity for developing resistance compared to classic antibiotics. While several AMPs have shown activity against antibiotic-sensitive and even multi-drug resistant strains, some bottlenecks to further development and clinical applications are still present, for instance, low antimicrobial activity, instability under physiological conditions, systemic toxicity and the potential for compromising the innate host defense immunity. Conjugation to molecules such as proteins, synthetic polymers, small molecules and nanoparticles are strategies under investigation to boost the therapeutic efficacy of AMPs. This review focuses on the design and application of AMPs’ conjugates. In silico tools for creating new AMPs and AMPs’ conjugates and their clinical development are also discussed. Furthermore, key future considerations regarding the major achievements and challenges of AMPs’ conjugates in the antimicrobial resistance context are presented as a take-home message.
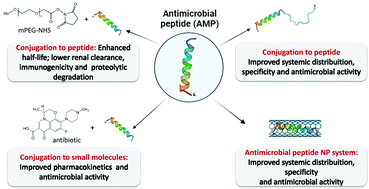
- This article is part of the themed collections: Celebrating International Women’s Day: Women in Materials Science and 2022 Journal of Materials Chemistry B Most Popular Articles
 Please wait while we load your content...
Please wait while we load your content...

