
Electrodeposited nickel-sulfide films as competent hydrogen evolution catalysts in neutral water†
Abstract
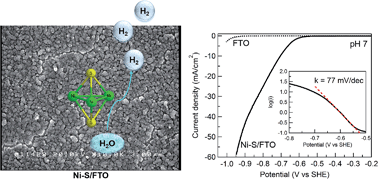
The development of low-cost, efficient, and robust electrocatalysts of the hydrogen evolution reaction (HER) is a crucial step toward the conversion and storage of sustainable and carbon-neutral energy resources, such as solar energy. Not only the HER catalysts need to be composed of inexpensive elements, they are also desirable to be prepared at low energy cost. In this work, we report that nickel-sulfide (Ni-S) films prepared by facile potentiodynamic deposition are active HER catalysts in aqueous media. Notably, the Ni-S films showed catalytic activity in water with a wide range of pH values (0 to 14), as well as in natural water. In pH 7 phosphate buffer, a current density of 60 mA cm−2 could be achieved with a Tafel slope of 77 mV dec−1 and a Faradaic efficiency of 100%. A long-term bulk electrolysis of the Ni-S film exhibited steady current over 100 h with no deactivation, demonstrating its superior stability in neutral water. Further, an initial activation process was observed, which is likely due to the increase in the effective surface area of the Ni-S film under electrocatalytic conditions. A suite of characterization techniques, including X-ray photoelectron spectroscopy and X-ray absorption spectroscopy, were conducted to probe the composition and structure of the Ni-S film, revealing that its major component is Ni3S2 which was preserved under electrocatalytic conditions.
 Please wait while we load your content...
Please wait while we load your content...

