
Cascade reactions of glycine Schiff bases and chiral phase transfer catalysts in the synthesis of α-amino acids 3-substituted phthalides or isoindolinones†
Abstract
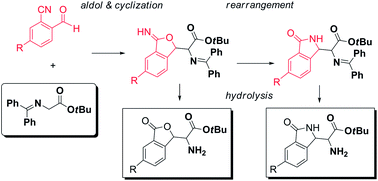
The tuning of aldol/cyclization cascade reactions of glycine Schiff bases with 2-cyano benzaldehydes provides access to non-natural α-amino acid derivatives substituted alternatively with phthalides or isoindolinones, depending on the strength of the used base. Moreover, a preliminary screening of catalysts and conditions for development of asymmetric versions identified chiral bifunctional phase transfer catalysts as particularly promising, leading to the α-amino esters 3-substituted phthalides in high yields and good diastereo- and enantioselectivity.
 Please wait while we load your content...
Please wait while we load your content...

