
Conquering three-carbon axial chirality of allenes†
Abstract
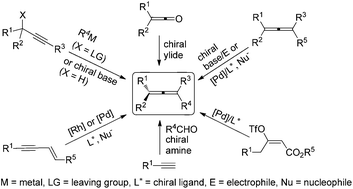
While one-carbon central chirality of organic molecules has been recognized and extensively studied for more than a century, far less attention has been paid to three-carbon axial chirality of allenes, although they exist in nature with interesting biological activity and have been demonstrated with great synthetic potentials. However, remarkable progress has been made in this field in recent years, giving rise to axially chiral allenes with a wide range of functionalities with practical enantioselectivity. This review provides a concise account of enantioselective syntheses of axially chiral allenes with a selection of published protocols.
- This article is part of the themed collections: Celebrating 70 Years of Shanghai Institute of Organic Chemistry and Celebrating the 80th Birthday of Professor Ei-ichi Negishi
 Please wait while we load your content...
Please wait while we load your content...

