
Copper-mediated difluoromethylation of electron-poor aryl iodides at room temperature†
Abstract
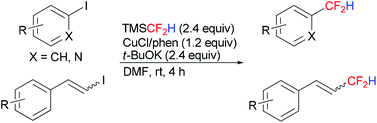
A convenient copper-mediated direct difluoromethylation of electron-deficient aryl iodides, as well as heteroaryl and β-styryl iodides, using TMSCF2H has been developed. This one-step protocol proceeded at room temperature, affording various difluoromethylated products in moderate to excellent yields.
- This article is part of the themed collection: Celebrating 70 Years of Shanghai Institute of Organic Chemistry
 Please wait while we load your content...
Please wait while we load your content...

