
Hydration of porphyrin and Mg–porphyrin: ab initio quantum mechanical charge field molecular dynamics simulations
Abstract
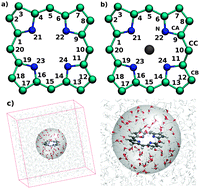
Ab initio QMCF-MD simulations were performed for porphyrin (POR) and magnesium–porphyrin (Mg–POR) immersed in water to study their structural and dynamical properties. The observed hydration behaviour of these solutes representing biomimetic models is in fair agreement with structural and dynamical features of their biological analogues, protoporphyrin IX (PPIX) and chlorophyll (CHl). Structural data obtained from the radial, angular and spatial distribution functions as well as the angular–radial distributions have a consensus on possessing a contrasting hydration behaviour of POR and Mg–POR. Flexibility of the ring in both solutes described by the improper torsional distribution and root mean square fluctuation showed an influence on H-bond interactions between the nitrogen atoms and water molecules that are also reflected in the respective dynamics. An axial water molecule coordinated to the Mg(II) ion indicates the penta-coordinated Mg–POR to be stable along the simulation. It was also shown that complexation of the Mg(II) ion to the porphyrin influences the hydration patterns significantly compared to the porphyrin itself, which is further supported by the vibrational power spectra evaluated for both solutes. Free energy of binding and solvent accessible surface area calculations also confirmed that these two solutes have distinct hydration behaviour. Detailed knowledge of the individual hydration patterns is expected to be of particular benefit.
 Please wait while we load your content...
Please wait while we load your content...