
A theoretical analysis of the phosphorescence efficiencies of Cu(i) complexes†
Abstract
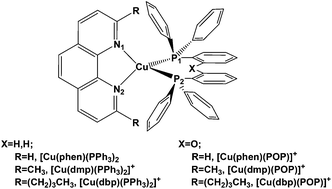
We herein report a theoretical analysis using density functional theory (DFT) and time-dependent DFT (TDDFT) to study the electronic structures and photophysical properties of mixed-ligand Cu(I) complexes. An evaluation of the non-radiative and radiative decay rate constants (knr and kr) is presented. It is found that large spin–orbit coupling (SOC) matrix elements do not necessarily result in large values of kr. Introducing the POP (bis[2-(diphenylphosphino)phenyl]ether) ligand instead of a pair of PPh3 (triphenylphosphine) ligands, it is found that the ether linkage plays an important role in governing the quantum efficiency of the studied complexes. However, the balance between hole injection and electron acceptance, which leads to the quantum yield of [Cu(dmp)(POP)]+ being close to that of [Cu(dbp)(POP)]+, is another important factor in tuning the quantum efficiency. A thorough understanding of the effect of the coordinating ligand on the photophysical behavior of a transition metal complex is desirable for the rational synthesis of highly phosphorescent materials.
- This article is part of the themed collection: Synergy between Experiment and Theory
 Please wait while we load your content...
Please wait while we load your content...

