
Symmetry matters: photodissociation dynamics of symmetrically versus asymmetrically substituted phenols†
Abstract
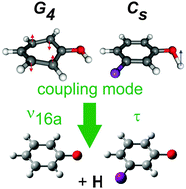
We report a combined experimental (H (Rydberg) atom photofragment translational spectroscopy) and theoretical (ab initio electronic structure and vibronic coupling calculations) study of the effects of symmetry on the photodissociation dynamics of phenols. Ultraviolet photoexcitation to the bound S1(1ππ*) state of many phenols leads to some O–H bond fission by tunneling through the barrier under the conical intersection (CI) between the S1 and dissociative S2(1πσ*) potential energy surfaces in the RO–H stretch coordinate. Careful analysis of the total kinetic energy release spectra of the resulting products shows that the radicals formed following S1 ← S0 excitation of phenol and symmetrically substituted phenols like 4-fluorophenol all carry an odd number of quanta in vibrational mode ν16a, whereas those deriving from asymmetrically substituted systems like 3-fluorophenol or 4-methoxyphenol do not. This contrasting behavior can be traced back to symmetry. Symmetrically substituted phenols exist in two equivalent rotamers, which interconvert by tunneling through the barrier to OH torsional motion. Their states are thus best considered in the non-rigid G4 molecular symmetry group, wherein radiationless transfer from the S1 to S2 state requires a coupling mode of a2 symmetry. Of the three a2 symmetry parent modes, the out-of-plane ring puckering mode ν16a shows much the largest interstate coupling constant in the vicinity of the S1/S2 CI. The nuclear motions associated with ν16a are orthogonal to the dissociation coordinate, and are thus retained in the radical products. Introducing asymmetry (even a non-linear substituent in the 4-position) lifts the degeneracy of the rotamers, and lowers the molecular symmetry to Cs. Many more parent motions satisfy the reduced (a′′) symmetry requirement to enable S1/S2 coupling, the most effective of which is OH torsion. This motion ‘disappears’ on O–H bond fission; symmetry thus imposes no restriction to forming radical products with vibrational quantum number v = 0. The present work yields values for the O–H bond strengths in 3-FPhOH and 4-MeOPhOH, and recommends modest revisions to the previously reported O–H bond strengths in other asymmetrically substituted phenols like 3- and 2-methylphenol and 4-hydroxyindole.
- This article is part of the themed collection: Imaging molecular dynamics
 Please wait while we load your content...
Please wait while we load your content...

