
Facile synthesis of N-aryl phenothiazines and phenoxazines via Brønsted acid catalyzed C–H amination of arenes†
Bin
Tan  *a
*a
*a
Abstract
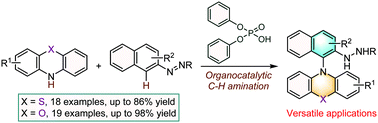
N-Aryl phenothiazines and phenoxazines are of significant importance in various disciplines throughout academia and industry. The conventional synthetic strategy for the construction of these structures centers on the transition-metal-catalyzed cross-coupling of aryl halides with phenothiazines or phenoxazines. Here we present an organocatalytic approach to access N-naphthyl phenothiazine and phenoxazine scaffolds through a straightforward C–H amination of arenes as enabled by an azo group. This reaction features operational simplicity, adequate substrate generality and excellent functional group compatibility. Notably, the efficiency of the catalyst could be perfectly preserved after 5 catalytic cycles.
 Please wait while we load your content...
Please wait while we load your content...

