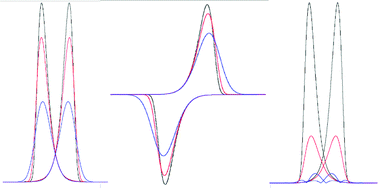
The potential-dependences of the rate constants associated with heterogeneous electron transfer predicted by the empirically based Butler–Volmer and fundamentally based Marcus–Hush formalisms are well documented for dc cyclic voltammetry. However, differences are often subtle, so, presumably on the basis of simplicity, the Butler–Volmer method is generally employed in theoretical–experimental comparisons. In this study, the ability of Large Amplitude Fourier Transform AC Cyclic Voltammetry to distinguish the difference in behaviour predicted by the two formalisms has been investigated. The focus of this investigation is on the difference in the profiles of the first to sixth harmonics, which are readily accessible when a large amplitude of the applied ac potential is employed. In particular, it is demonstrated that systematic analysis of the higher order harmonic responses in suitable kinetic regimes provides predicted deviations of Marcus–Hush from Butler–Volmer behaviour to be established from a single experiment under conditions where the background charging current is minimal.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


