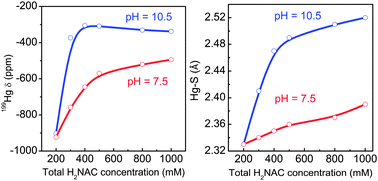
N-Acetylcysteine (H2NAC) is a potent antioxidant, a precursor for cysteine and glutathione, and a potential antidote against certain metal ions such as cadmium and mercury. Little is known about the structural aspects of complexes formed between Hg(II) and N-acetylcysteine, despite many biological tests on its ability to bind to organic and inorganic mercury, and a few reports on formation constants for Hg(NAC)n (n = 1–3) complexes. We have combined several techniques, including Hg L3-edge EXAFS (extended X-ray absorption fine structure), 199Hg NMR and Raman spectroscopy, to investigate the nature and structure of Hg(II) N-acetylcysteine complexes formed in aqueous solution at pH 7.5 and 10.5. To allow measurements on the same samples, rather concentrated solutions containing CHg(II) = 0.1 M and variable H2NAC/Hg(II) mole ratios = 2.0–10.0 were used. At physiological pH, Hg(NAC)22− and Hg(NAC)34− complexes form, while in ligand excess and at alkaline pH (H2NAC/Hg(II) > 4), a novel tetra-thiolate species Hg(NAC)46− dominates. Comparison between the Hg(II) complex formation with cysteine, penicillamine and N-acetylcysteine in alkaline aqueous solution has been made to elucidate the influence of the blocked amino group of N-acetylcysteine.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?
