
Progress in redox flow batteries, remaining challenges and their applications in energy storage
Abstract
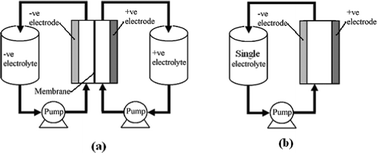
Redox flow batteries, which have been developed over the last 40 years, are used to store energy on the medium to large scale, particularly in applications such as load levelling, power quality control and facilitating renewable energy deployment. Various
- This article is part of the themed collection: Battery development over the last decade
 Please wait while we load your content...
Please wait while we load your content...

