
Predictive methods for the estimation of thermophysical properties of ionic liquids
Abstract
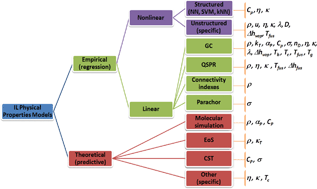
While the design of products and processes involving ionic liquids (ILs) requires knowledge of the thermophysical properties for these compounds, the massive number of possible distinct ILs precludes their detailed experimental characterization. To overcome this limitation, chemists and engineers must rely on predictive models that are able to generate reliable values for these properties, from the knowledge of the structure of the IL. A large body of literature was developed in the last decade for this purpose, aiming at developing predictive models for thermophysical and transport properties of ILs. A critical review of those models is reported here. The modelling approaches are discussed and suggestions relative to the current best methodologies for the prediction of each property are presented. Since most of the these works date from the last 5 years, this field can still be considered to be in its infancy. Consequently, this work also aims at highlighting major gaps in both existing data and modelling approaches, identifying unbeaten tracks and promising paths for further development in this area.
- This article is part of the themed collection: Machine learning and artificial neural networks in chemistry
 Please wait while we load your content...
Please wait while we load your content...

