
Convergence of QM/MM free-energy perturbations based on molecular-mechanics or semiempirical simulations
Abstract
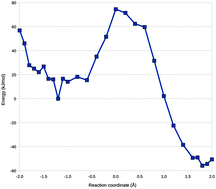
Lately, there has been great interest in performing free-energy perturbation (FEP) at the combined quantum mechanics and molecular mechanics (QM/MM) level, e.g. for enzyme reactions. Such calculations require extensive sampling of phase space, which typically is prohibitive with density-functional theory or ab initio methods. Therefore, such calculations have mostly been performed with semiempirical QM (SQM) methods, or by using a thermodynamic cycle involving sampling at the MM level and perturbations between the MM and QM/MM levels of theory. However, the latter perturbations typically have convergence problems, unless the QM system is kept fixed during the simulations, because the MM and QM/MM descriptions of the internal degrees of freedom inside the QM system are too dissimilar. We have studied whether the convergence of the MM → QM/MM perturbation can be improved by using a thoroughly parameterised force field or by using SQM/MM methods. As a test case we use the first half-reaction of
- This article is part of the themed collection: Theoretical chemical physics of biological systems
 Please wait while we load your content...
Please wait while we load your content...

