
Sevenfold enhancement on porphyrindye efficiency by coordination of ruthenium polypyridine complexes†‡
Abstract
Sevenfold enhancement of photoconversion efficiency was achieved by incorporation of peripheral ruthenium complexes to a
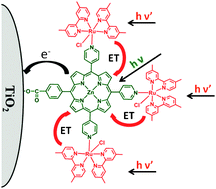
- This article is part of the themed collection: Porphyrins & Phthalocyanines
 Please wait while we load your content...
Please wait while we load your content...

