
Two-dimensional carbide/nitride (MXene) materials in thermal catalysis
 *a
*a
Abstract
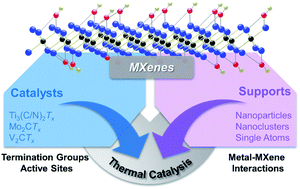
Two-dimensional carbide/nitride materials, i.e., MXenes, were first synthesized from the corresponding MAX phases in 2011. Since their discovery, they have been widely applied in batteries, supercapacitors, electromagnetic shielding materials, electrocatalysis, and photocatalysis due to their unique and tunable physical, chemical, and electrical properties. Recently, MXenes have been applied in thermal catalytic reactions, such as hydrogenation, dehydrogenation, water–gas shift reactions, and desulfurization due to their thermal stability and superior catalyst properties similar to noble metals. In this article, we systematically summarize the characteristics of MXenes as catalysts and supports compared with other traditional thermal catalysts with respect to both structures and catalytic activities. Furthermore, the nature of termination groups, active sites, and metal–MXene interactions are elaborated. Finally, we provide insights into the future development of catalysts based on MXene materials.
- This article is part of the themed collection: Journal of Materials Chemistry A Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

