
The role of electrostatic induction in secondary isotope effects on acidity: theory and computational confirmation†
Abstract
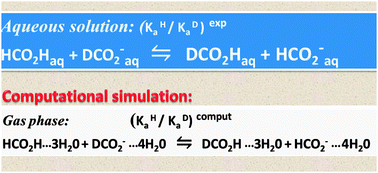
The role of electrostatic induction in secondary isotope effects (SIEs) on acidity is reconsidered. A first order perturbation treatment demonstrates that an electric charge acting on a C–H bond vibrating in its anharmonic potential induces a mass-dependent change in its harmonic force constant and – accordingly – an isotope-dependent shift of its zero-point vibrational energy (ZPVE). Its sense and magnitude are determined by the bond-dipole derivative, dμ/dr. The SIE on the dissociation equilibrium of formic acid was evaluated by gas-phase computations (MP2/311G**) on formic acid and the formate ion, bare and variously hydrated. The CH-stretching frequencies, as well as the IEs on the ZPVE and free energy of ionization (ΔG0298), are in linear correlation with Δμ/Δre, the ratio of the change in the C–H bond moment upon protonation to the corresponding equilibrium bond length difference, suggesting that Δμ/Δre is an adequate linear approximation to dμ/dr and confirming the electrostatic origin of the isotope effect. Similar computations on the dissociation of acetic acid and methylammonium ions yield analogous results. In both cases, the mean equilibrium length of the methyl CH bonds, rm, depends strongly on μg, the CH3 group dipole moment and, when allowance is made for steric effects, the IEs on ZPVE and ΔG0298 are in linear correlation with μg. In each of the three equilibria, the isotopic ΔpKa value computed for the polyhydrated acid and conjugate base is smaller – in the case of the carboxylic acids much smaller – than that computed with the bare species, reproducing the published experimental values in aqueous solution to within a factor of two.
 Please wait while we load your content...
Please wait while we load your content...

