
A QM/MM study of the initial excited state dynamics of green-absorbing proteorhodopsin†

Abstract
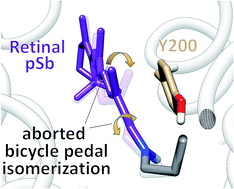
The primary photochemical reaction of the green-absorbing proteorhodopsin is studied by means of a hybrid quantum mechanics/molecular mechanics (QM/MM) approach. The simulations are based on a homology model derived from the blue-absorbing proteorhodopsin crystal structure. The geometry of retinal and the surrounding sidechains in the protein binding pocket were optimized using the QM/MM method. Starting from this geometry the isomerization was studied with a relaxed scan along the C13![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C14 dihedral. It revealed an “aborted bicycle pedal” mechanism of isomerization that was originally proposed by Warshel for bovine rhodopsin and bacteriorhodopsin. However, the isomerization involved the concerted rotation about C13C14 and C15N, with the latter being highly twisted but not isomerized. Further, the simulation showed an increased steric interaction between the hydrogen at the C14 of the isomerizing bond and the hydroxyl group at the neighbouring tyrosine 200. In addition, we have simulated a nonadiabatic trajectory which showed the timing of the isomerization. In the first 20 fs upon excitation the order of the conjugated double and single bonds is inverted, consecutively the C13C14 rotation is activated for 200 fs until the S1–S0 transition is detected. However, the isomerization is reverted due to the specific interaction with the tyrosine as observed along the relaxed scan calculation. Our simulations indicate that the retinal – tyrosine 200 interaction plays an important role in the outcome of the photoisomerization.
C14 dihedral. It revealed an “aborted bicycle pedal” mechanism of isomerization that was originally proposed by Warshel for bovine rhodopsin and bacteriorhodopsin. However, the isomerization involved the concerted rotation about C13C14 and C15N, with the latter being highly twisted but not isomerized. Further, the simulation showed an increased steric interaction between the hydrogen at the C14 of the isomerizing bond and the hydroxyl group at the neighbouring tyrosine 200. In addition, we have simulated a nonadiabatic trajectory which showed the timing of the isomerization. In the first 20 fs upon excitation the order of the conjugated double and single bonds is inverted, consecutively the C13C14 rotation is activated for 200 fs until the S1–S0 transition is detected. However, the isomerization is reverted due to the specific interaction with the tyrosine as observed along the relaxed scan calculation. Our simulations indicate that the retinal – tyrosine 200 interaction plays an important role in the outcome of the photoisomerization.
- This article is part of the themed collection: Photoinduced Processes in Nucleic Acids and Proteins
 Please wait while we load your content...
Please wait while we load your content...

