
Density functional theory analysis of structural and electronic properties of orthorhombic perovskite CH3NH3PbI3
Abstract
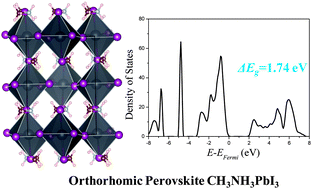
The organic–inorganic hybrid perovskite CH3NH3PbI3 is a novel light harvester, which can greatly improve the solar-conversion efficiency of dye-sensitized solar cells. In this article, a first-principle theoretical study is performed using local, semi-local and non-local exchange–correlation approximations to find a suitable method for this material. Our results, using the non-local optB86b + vdWDF functional, excellently agree with the experimental data. Thus, consideration of weak van der Waals interactions is demonstrated to be important for the accurate description of the properties of this type of organic–inorganic hybrid materials. Further analysis of the electronic properties reveals that I 5p electrons can be photo-excited to Pb 6p empty states. The main interaction between the organic cations and the inorganic framework is through the ionic bonding between CH3 and I ions. Furthermore, I atoms in the Pb–I framework are found to be chemically inequivalent because of their different chemical environments.
 Please wait while we load your content...
Please wait while we load your content...

