
Singlet and triplet excitations in chemically functionalized single-walled carbon nanotubes
 *ae
Yulun
Han,
be
Grace
Tiffany,
de
Dmitri
Kilin,
e
Sergei
Tretiak
c
and
Svetlana
Kilina
*e
*ae
Yulun
Han,
be
Grace
Tiffany,
de
Dmitri
Kilin,
e
Sergei
Tretiak
c
and
Svetlana
Kilina
*e
Abstract
We investigate the lowest-energy singlet (S1) and triplet (T1) excitons in covalently functionalized (11,0) single-walled carbon nanotubes (SWCNTs) using spin-polarized constrained – occupation density functional theory (CO-DFT) under periodic boundary conditions. Our study focuses on how the singlet–triplet energy splitting (ΔEST) and exciton localization are influenced by the position of the sp3 -defects formed by aryl-based adducts with varying electron-donating and electron-withdrawing abilities. We find that the PBE functional within CO-DFT reliably predicts the optical redshift of defect-related singlet excitons 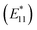 compared to the main-band E11 of the pristine nanotube, matching experimental values better than B3LYP. Across all studied defect configurations, the T1 state consistently lies below S1, and we observe a strong correlation between ΔEST and the optical redshift: defect configurations inducing larger redshifts also result in greater ΔEST, ranging from 6 to 36 meV. Although the molecular adduct has a minor effect on the ΔEST energy, aryl with strong electron-withdrawing groups, such as NO2, significantly enhances the charge-transfer character of both S1 and T1 by partially localizing electron density on the molecule, while the hole is located around the defect site. These adducts also reduce geometric and electronic reorganization between S1 and T1 excitons. Both properties are the key factors that are known to support thermally activated delayed fluorescence (TADF) owing to a smaller value of ΔEST. Altogether, our results predict electron-withdrawing functional groups as an effective strategy for engineering TADF in SWCNTs, positioning these systems as promising near-infrared emitters for quantum light sources, spin-based devices, and energy-efficient optoelectronic applications.
compared to the main-band E11 of the pristine nanotube, matching experimental values better than B3LYP. Across all studied defect configurations, the T1 state consistently lies below S1, and we observe a strong correlation between ΔEST and the optical redshift: defect configurations inducing larger redshifts also result in greater ΔEST, ranging from 6 to 36 meV. Although the molecular adduct has a minor effect on the ΔEST energy, aryl with strong electron-withdrawing groups, such as NO2, significantly enhances the charge-transfer character of both S1 and T1 by partially localizing electron density on the molecule, while the hole is located around the defect site. These adducts also reduce geometric and electronic reorganization between S1 and T1 excitons. Both properties are the key factors that are known to support thermally activated delayed fluorescence (TADF) owing to a smaller value of ΔEST. Altogether, our results predict electron-withdrawing functional groups as an effective strategy for engineering TADF in SWCNTs, positioning these systems as promising near-infrared emitters for quantum light sources, spin-based devices, and energy-efficient optoelectronic applications.

 Please wait while we load your content...
Please wait while we load your content...

