
A DFT analysis of structural and electronic modulation of Cs2AgBiX6 (X = Cl, Br) via A-site NH4+ substitution for photovoltaic applications

Abstract
To address environmental pollution and sustainable energy challenges, lead-free Ag–Bi double perovskites Cs2AgBiX6 (X = Cl, Br) and their ammonium-substituted variants CsNH4AgBiX6 and (NH4)2AgBiX6 are investigated using first-principles FP-LAPW calculations within density functional theory. Ammonium incorporation slightly reduces lattice size while enhancing structural flexibility. Band-structure analysis (GGA, SOC, hybrid-PBE) shows decreasing band gaps with NH4 doping from 2.52 eV to 2.09 eV, with the CBM dominated by Bi states and the VBM by halide p states. Effective mass calculations indicate high carrier mobility due to the low effective mass of (NH4)2AgBiX6 (X = Cl, Br) compared to Cs-based double perovskites, which results in 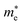 values that are between 0.524 and 0.939 eV, and
values that are between 0.524 and 0.939 eV, and 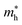 values that are between 1.2 and 1.645 eV. The stability of these compounds is confirmed through mechanical (Cij), formation of enthalpy ΔHf and Goldschmidt tolerance factor (τG) analyses. The elastic constants confirm the mechanically stable and ductile nature of these materials. Furthermore, ab initio molecular dynamics simulations and phonon band-structure calculations have been performed and confirm the stability of the materials. Optical properties reveal stronger light absorption (∼45 × 104 cm−1 in the visible region) and an enhanced dielectric response after NH4+ substitution. Band-edge alignment analysis supports the potential for photocatalytic water splitting, while SLME analysis identifies (NH4)2AgBiBr6 (ηmax = 6.47%) as the most promising photovoltaic absorber. Overall, A-site ammonium engineering effectively tunes the structural, electronic, optical, and photocatalytic properties of Ag–Bi double perovskites for energy applications.
values that are between 1.2 and 1.645 eV. The stability of these compounds is confirmed through mechanical (Cij), formation of enthalpy ΔHf and Goldschmidt tolerance factor (τG) analyses. The elastic constants confirm the mechanically stable and ductile nature of these materials. Furthermore, ab initio molecular dynamics simulations and phonon band-structure calculations have been performed and confirm the stability of the materials. Optical properties reveal stronger light absorption (∼45 × 104 cm−1 in the visible region) and an enhanced dielectric response after NH4+ substitution. Band-edge alignment analysis supports the potential for photocatalytic water splitting, while SLME analysis identifies (NH4)2AgBiBr6 (ηmax = 6.47%) as the most promising photovoltaic absorber. Overall, A-site ammonium engineering effectively tunes the structural, electronic, optical, and photocatalytic properties of Ag–Bi double perovskites for energy applications.

 Please wait while we load your content...
Please wait while we load your content...

