
DFT insights into the stability of single-metal-atoms on Mo-based o-MXenes driven by the ligand effect

Abstract
MXenes, a novel class of two-dimensional transition metal carbides, nitrides or carbonitrides, have emerged as promising supports for single-atom catalysts owing to their tunable structural and electronic properties. Using density functional theory calculations, we investigate ordered Mo-based bimetallic MXenes without surface functional groups (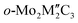 and o-Mo2M″C2, where M″ = Ti, Zr, Hf, V, Nb and Ta) as materials to stabilize platinum-group-metal (PGM) single atoms at Mo vacancies. Compared with the corresponding Mo-only MXenes, introducing a second metal component, M″, strengthens PGM anchoring and increases sintering resistance, which we attributed to a ligand effect that enhances PGM-C covalency and reorganizes the PGM d states. This stabilization is further captured by a combined electronic descriptor δ = εp − εd. Here, δ is defined as the difference between the p band center (εp) of the C atoms adjacent to the PGM and the d band center (εd) of the PGM atom. This descriptor δ shows a strong correlation with the binding strength between PGM and MXenes. To assess the changes of PGM reactivity raised by different MXenes, we use CO, CH3 and NH3 adsorption strength, highlighting a stability-reactivity trend. An XGBoost-based machine learning analysis further revealed that adsorption energies are governed by multiple electronic descriptors (beyond εd), with the average electronegativity (χavg) of the PGM-MXene system contributing to the observed trends in adsorption energies. This study provides a new atomic-level understanding of the rational design principle for stabilizing atomically dispersed SACs on MXenes and provides guidelines for the experimental synthesis.
and o-Mo2M″C2, where M″ = Ti, Zr, Hf, V, Nb and Ta) as materials to stabilize platinum-group-metal (PGM) single atoms at Mo vacancies. Compared with the corresponding Mo-only MXenes, introducing a second metal component, M″, strengthens PGM anchoring and increases sintering resistance, which we attributed to a ligand effect that enhances PGM-C covalency and reorganizes the PGM d states. This stabilization is further captured by a combined electronic descriptor δ = εp − εd. Here, δ is defined as the difference between the p band center (εp) of the C atoms adjacent to the PGM and the d band center (εd) of the PGM atom. This descriptor δ shows a strong correlation with the binding strength between PGM and MXenes. To assess the changes of PGM reactivity raised by different MXenes, we use CO, CH3 and NH3 adsorption strength, highlighting a stability-reactivity trend. An XGBoost-based machine learning analysis further revealed that adsorption energies are governed by multiple electronic descriptors (beyond εd), with the average electronegativity (χavg) of the PGM-MXene system contributing to the observed trends in adsorption energies. This study provides a new atomic-level understanding of the rational design principle for stabilizing atomically dispersed SACs on MXenes and provides guidelines for the experimental synthesis.

 Please wait while we load your content...
Please wait while we load your content...

