
A structural similarity based data-mining algorithm for modeling multi-reactant heterogeneous catalysts†

Abstract
First-principles-based Density Functional Theory (DFT) simulations are powerful tools for studying heterogeneous catalyst systems. However, their high computational cost and large configuration space hinder their application in understanding multi-reactant catalysis on geometrically diverse surfaces. This work introduces an innovative similarity algorithm that quantifies the structural differences between atomic configurations to address this challenge. The quantification effectively identifies structurally dissimilar configurations with minimal human intervention. Consequently, data mining the configurational phase-space through this similarity algorithm drastically reduces the number of DFT simulations required to identify stable atomic models relevant to key multi-reactant chemistries. In this work, the similarity algorithm is utilized to understand CO*–OH* co-adsorption at varying adsorbate coverages on a stepped Pt surface by DFT simulating only 2% of possible unique configurations. Furthermore, the versatility of the similarity algorithm is showcased by analyzing bidentate adsorption 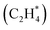 on a stepped Pt surface. This work serves as a crucial steppingstone towards understanding important multi-reactant heterogeneous catalytic chemistries.
on a stepped Pt surface. This work serves as a crucial steppingstone towards understanding important multi-reactant heterogeneous catalytic chemistries.

 Please wait while we load your content...
Please wait while we load your content...

