
Selectivity and activity modulation of electrocatalytic carbon dioxide reduction by atomically dispersed dual iron catalysts†

Abstract
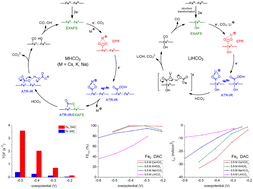
By regulating the electronic environment of Fe active centers to modulate electrocatalytic CO2 reduction behavior, an advanced dual Fe2-site catalyst (Fe2 DAC) exhibiting CO current density (jCO) of 10 mA cm−2 at an overpotential of 330 mV in a CO2-saturated 0.5 M KHCO3 electrolyte was designed and characterized by HAADF-STEM microscopy and XAS/EPR spectroscopies. With regard to Fe2 DAC displaying a higher charge transfer coefficient (α = 0.53), turnover frequency (TOFCO = 2.03 s−1), and CO faradaic efficiency (FECO = 98.6%) at an overpotential of 400 mV compared to that of single iron atom catalyst (Fe SAC, α = 0.31, TOFCO = 0.25 s−1, FECO = 60.1%), the kinetic mechanism was investigated/elucidated by the cation effect and in operando spectroscopy. The higher DMPO–CO2 EPR intensity (g = 2.0065, aN = 15.6 G, aH = 18.9 G) and the smaller separation of ATR-SEIRAS stretching frequencies (1554 (νasym), 1288 (νsym) cm−1, Δν = 266 cm−1) suggest that the structural type of the [*COOCs]˙/[*COOCs]− intermediate is μ2–η3 CO2 coordination (class II) for Fe2 DAC-triggered electrocatalytic CO2 reduction in CO2-saturated CsHCO3 solution. The strong orbital interaction among the dual Fe2 site, intermediate [CO2]˙−/[CO2]2−, and Cs+ cation (6s orbital) is proposed to accelerate charge transfer kinetics and shift the rate-determining step from the electron transfer step (Li+, Na+) to the protonation step (K+, Cs+), as evidenced by Cs+-induced increase in the proton reaction order (0.86) and Cs+-induced decrease in the kinetic Tafel slope (57.2 mV dec−1) and electrochemical activation energy (23.7 kJ mol−1). In contrast, the structural transformation from the dual FeII2 motif to a single FeII site revealed by the disappearance of the Fe–Fe distance (3.10 Å) in operando Fe K edge EXAFS lends support to the absence of stretching frequencies (1429 (νasym), 1380 (νasym), 1241 (νasym) cm−1) ascribed to μ2–η2 CO2 coordination in a CO2-saturated LiHCO3 aqueous medium, demonstrating that the transformation of [*COOCs]−/[*COOK]− into the bridge [CO2]2− [Fe–μ-C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)O–Fe] is vital for electrocatalytic CO2-to-CO conversion. In addition to identifying the dinuclear Fe2II site as a catalytic center, this study demonstrates that the thermodynamic stabilization effect of both the cation size (large s orbital/soft hydration shell) and dual Fe2II motif toward [CO2]˙−/[CO2]2− intermediate is pivotal to the superior CO2RR kinetics (activity/selectivity). The proposed pathways (Cs+/K+/Na+vs. Li+ and dual Fe2 site vs. single Fe site) may provide insights into how the orbital interaction and the peculiar electronic structure of the dinuclear Fe2 site impacts the molecular-level mechanism for efficient electrocatalytic CO2 reduction.
O)O–Fe] is vital for electrocatalytic CO2-to-CO conversion. In addition to identifying the dinuclear Fe2II site as a catalytic center, this study demonstrates that the thermodynamic stabilization effect of both the cation size (large s orbital/soft hydration shell) and dual Fe2II motif toward [CO2]˙−/[CO2]2− intermediate is pivotal to the superior CO2RR kinetics (activity/selectivity). The proposed pathways (Cs+/K+/Na+vs. Li+ and dual Fe2 site vs. single Fe site) may provide insights into how the orbital interaction and the peculiar electronic structure of the dinuclear Fe2 site impacts the molecular-level mechanism for efficient electrocatalytic CO2 reduction.
 Please wait while we load your content...
Please wait while we load your content...

