
CrystalClear: an open, modular protocol for predicting molecular crystal growth from solution†

Abstract
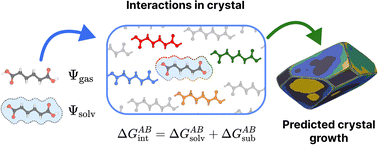
We present a new protocol for the prediction of free energies that determine the growth of sites in molecular crystals for subsequent use in Monte Carlo simulations using tools such as CrystalGrower [Hill et al., Chemical Science, 2021, 12, 1126–1146]. Key features of the proposed approach are that it requires minimal input, namely the crystal structure and solvent only, and provides automated, rapid generation of the interaction energies. The constituent components of this protocol, namely interactions between molecules (growth units) in the crystal, solvation contributions and treatment of long-range interactions are described in detail. The power of this method is shown via prediction of crystal shapes for ibuprofen grown from ethanol, ethyl acetate, toluene and acetonitrile, adipic acid grown from water, and five polymorphs (ON, OP, Y, YT04 and R) of ROY (5-methyl-2-[(2-nitrophenyl)amino]-3-thiophenecarbonitrile), with promising results. The predicted energies may be used directly or subsequently refined against experimental data, facilitating insight into the interactions governing crystal growth, while also providing a prediction of the solubility of the material. The protocol has been implemented in standalone, open-source software made available alongside this publication.
- This article is part of the themed collection: #MyFirstChemSci 2023
 Please wait while we load your content...
Please wait while we load your content...

