
Ir–Na cooperativity controls the diastereoselectivity of borrowing hydrogen C–C alkylation on isosorbide: synthesis methodology and mechanistic investigation†
 a
Marie-Ève L.
Perrin,
a
Maïwenn
Jacolot,
*a
Pierre-Adrien
Payard
*a
and
Florence
Popowycz
*a
a
Marie-Ève L.
Perrin,
a
Maïwenn
Jacolot,
*a
Pierre-Adrien
Payard
*a
and
Florence
Popowycz
*a
Abstract
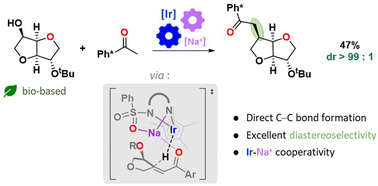
The direct functionalization of isosorbide is a current challenge of biomass valorisation due to the low selectivities usually obtained. Taking advantage of our preliminary observations concerning the diastereoselective Ir-catalyzed amination of isohexides, we report the first direct C–C borrowing hydrogen functionalisation on isosorbide. The C–C alkylation was achieved with excellent diastereocontrol using an Ir-based organometallic homogeneous catalyst. The reaction sequence involves a catalytic dehydrogenation that converts the alcohol into a reactive ketone, an in situ aldol condensation/crotonisation, and a catalytic dehydrogenation leading to the desired homologated product. Once the parameters that control the reaction outcome were explored and optimized, the stereoelectronic effects from which the diastereoselectivity originates were investigated by means of a computational mechanistic investigation performed at the DFT level. The roles played by the Na counter-cation to coordinate substrate's oxygen atoms and to guide hydride addition were evidenced allowing us to rationalize the experimental selectivity at the molecular level.
 Please wait while we load your content...
Please wait while we load your content...

