
Effective electronic tuning of Pt single atoms via heterogeneous atomic coordination of (Co,Ni)(OH)2 for efficient hydrogen evolution†
 ‡c
Lihua
Zhu,
*ab
Guoliang
Chai,
*cg
Qingsheng
Gao,
e
Bing Hui
Chen
f
and
Zhengxiao
Guo
*b
‡c
Lihua
Zhu,
*ab
Guoliang
Chai,
*cg
Qingsheng
Gao,
e
Bing Hui
Chen
f
and
Zhengxiao
Guo
*b
Abstract
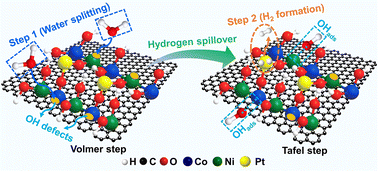
Hydroxide-supported atomic structures, particularly single atoms, offer a wide scope for active microenvironmental tuning to enhance the catalytic performance, but little has been explored on the electronic synergy between mono- and dual-hydroxides. Here, we propose a way of constructing a Pt1/(Co,Ni)(OH)2/C single-atom catalyst (SAC), with Pt single-atoms, Pt1, stabilized and anchored on the surface of defective (Co,Ni)(OH)2, which is further supported on carbon black. This catalyst exhibits a far superior hydrogen evolution reaction (HER) activity to Pt1/Co(OH)2/C, Pt1/Ni(OH)2/C, Pt1/C, and the commercial 20 wt% Pt/C. Particularly, it shows an almost zero onset overpotential and an outstanding electrocatalytic mass activity for the HER, 29.7 times higher than that of Pt1/C and 115.9 times higher than that of the commercial 20 wt% Pt/C at −0.09 V vs. RHE, respectively. There is negligible attenuation after the chronopotentiometry test at 100 mA cm−2 for 24 h and cyclic voltammetry for 20 000 cycles. Operando Raman spectra clarified that the Volmer step for water decomposition (the H–OH bond breaking) takes place around the defective sites of (Co,Ni)(OH)2. Density functional theory (DFT) calculations confirmed the electronic synergy between the Pt single atoms and bimetallic (Co,Ni)(OH)2, which leads to stable anchoring of Pt and yields an appropriate adsorption energy of *H, leading to rapid H2 generation.
 Please wait while we load your content...
Please wait while we load your content...

