
A computational mechanistic study of CH hydroxylation with mononuclear copper–oxygen complexes†

Abstract
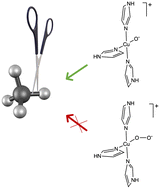
We present a computational study of methane hydroxylation by oxygen-bound monocopper complexes with the twin goals of resolving the active site identity and preferred mechanism of CH activation. Oxyl ([Cu(II)O(Im)3]+) and superoxo ([Cu(II)OO(Im)3]+) monocopper centers coordinated to three imidazole N-donors are investigated. Constrained density functional theory (CDFT) is necessary to overcome delocalization errors inherent to DFT and generate catalyst geometries with physically meaningful charge and spin at the active site. A comparison of ground-state triplet and excited-state singlet potential energy profiles for CH hydroxylation shows that oxyl is the more likely active site. For oxyl, spin crossing from the triplet to singlet potential energy surface may yield a lower energy hydroxylation pathway when compared to the pure triplet surface. The calculation of minimum energy crossing points is an essential next step towards determining whether CH hydroxylation proceeds via a two-step radical or single-step oxo-insertion mechanism.
- This article is part of the themed collection: Emerging Investigator Series
 Please wait while we load your content...
Please wait while we load your content...

