
First principles terahertz spectroscopy of molecular crystals: the crucial role of periodic boundary conditions benchmarked with experimental l-ascorbic acid spectra†
 *a
and
Wenyue
Guo
*a
*a
and
Wenyue
Guo
*a
Abstract
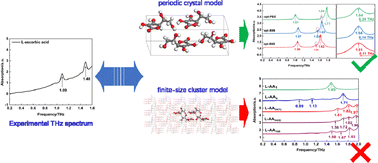
The terahertz (THz) region vibration spectral signatures of molecular crystals can usually be ascribed to the low-frequency vibrational modes related to weak intermolecular interactions, e.g. van der Waals (vdW) interactions or hydrogen bonding. These interactions collectively dictate the compositional units deviating from their equilibrium configurations. The collective movements are intrinsically long-range, and hence the boundary conditions used for theoretical calculation can affect the corresponding potential energy gradients and alter the vibrational features. In this work, we constructed a series of finite-sized cluster models with varying sizes and an extended periodic crystal model for L-ascorbic acid (L-AA) crystals. Density functionals with both semi-local contributions and nonlocal vdW terms, implemented with either atom-centered Gaussian basis or plane waves, were tested. By comparing first principles calculations with experimental time-domain spectra (TDS), we found that the non-local vdW functional opt-B88 combined with a periodic boundary condition is capable of assigning all the experimental features in the 0.2–1.6 THz region. Calculations with cluster models failed in this task. Even worse, the deficiency of the cluster models varied with cluster sizes, and did not converge as the cluster size grew. Our results demonstrate that an appropriate periodic boundary condition is essential to correctly assign and analyze the THz vibration spectra of molecular crystals.
 Please wait while we load your content...
Please wait while we load your content...

