
Efficient and accurate density-based prediction of macromolecular polarizabilities†

Abstract
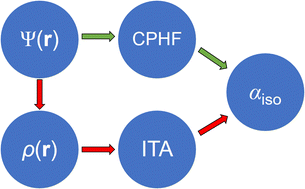
Accurately and efficiently predicting macromolecules’ polarizabilities is an open problem. In this work, we employ a few simple density-based quantities from the information-theoretic approach (ITA) to predict polarizability of proteins. We first build quantitative structure/property relationships between molecular polarizabilities and ITA quantities. We then verify the broad applicability of ITA quantities for polarizability prediction for inorganic, organic, and biological systems with both localized and delocalized electronic structure. As a proof-of-concept application, we predict the molecular polarizabilities of complex proteins. Based on the linear regression equations for 20 natural amino acid residues, 400 dipeptides, and 8000 tripeptides, one then predicts the molecular polarizability of a larger peptide or even a protein once the molecular wavefunction is obtained. Because it is extremely costly to determine the wavefunction for a macromolecule like a protein, we propose to combine the ITA with the linear-scaling generalized energy-based fragmentation (GEBF) method to predict the macromolecular polarizability. In GEBF, the total molecular polarizability is obtained as a linear combination of the corresponding quantities from a series of small subsystems. We can predict them based on the subsystem wavefunction and linear regression equations rather than compute them from the nearly-intractable coupled-perturbed Hartree–Fock or Kohn–Sham equations for the whole macromolecule. Computational results showcase that the GEBF-ITA protocol should be an inexpensive yet accurate theoretical tool for predicting macromolecular polarizabilities.
 Please wait while we load your content...
Please wait while we load your content...

