
Towards de novo design of transmembrane α-helical assemblies using structural modelling and molecular dynamics simulation
 *a
and
Yuji
Sugita
*abc
*a
and
Yuji
Sugita
*abc
Abstract
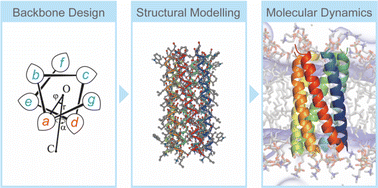
Computational de novo protein design involves iterative processes consisting of amino acid sequence design, structural modelling and scoring, and design validation by synthesis and experimental characterisation. Recent advances in protein structure prediction and modelling methods have enabled the highly efficient and accurate design of water-soluble proteins. However, the design of membrane proteins remains a major challenge. To advance membrane protein design, considering the higher complexity of membrane protein folding, stability, and dynamic interactions between water, ions, lipids, and proteins is an important task. For introducing explicit solvents and membranes to these design methods, all-atom molecular dynamics (MD) simulations of designed proteins provide useful information that cannot be obtained experimentally. In this review, we first describe two major approaches to designing transmembrane α-helical assemblies, consensus and de novo design. We further illustrate recent MD studies of membrane protein folding related to protein design, as well as advanced treatments in molecular models and conformational sampling techniques in the simulations. Finally, we discuss the possibility to introduce MD simulations after the existing static modelling and screening of design decoys as an additional step for refinement of the design, which considers membrane protein folding dynamics and interactions with explicit membranes.
- This article is part of the themed collections: 2023 PCCP HOT Articles and 2023 PCCP Reviews
 Please wait while we load your content...
Please wait while we load your content...

