
Learning molecular dynamics: predicting the dynamics of glasses by a machine learning simulator†

Abstract
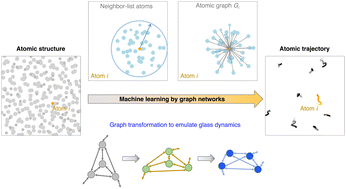
Many-body dynamics of atoms such as glass dynamics is generally governed by complex (and sometimes unknown) physics laws. This challenges the construction of atom dynamics simulations that both (i) capture the physics laws and (ii) run with little computation cost. Here, based on graph neural network (GNN), we introduce an observation-based graph network (OGN) framework to “bypass all physics laws” to simulate complex glass dynamics solely from their static structure. By taking the example of molecular dynamics (MD) simulations, we successfully apply the OGN to predict atom trajectories evolving up to a few hundred timesteps and ranging over different families of complex atomistic systems, which implies that the atom dynamics is largely encoded in their static structure in disordered phases and, furthermore, allows us to explore the capacity of OGN simulations that is potentially generic to many-body dynamics. Importantly, unlike traditional numerical simulations, the OGN simulations bypass the numerical constraint of small integration timestep by a multiplier of ≥5 to conserve energy and momentum until hundreds of timesteps, thus leapfrogging the execution speed of MD simulations for a modest timescale.
- This article is part of the themed collection: Machine Learning and Artificial Intelligence: A cross-journal collection
 Please wait while we load your content...
Please wait while we load your content...

