
High-throughput computational workflow for ligand discovery in catalysis with the CSD†

Abstract
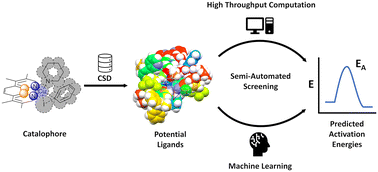
A novel semi-automated, high-throughput computational workflow for ligand/catalyst discovery based on the Cambridge Structural Database is reported. Two potential transition states of the Ullmann–Goldberg reaction were identified and used as a template for a ligand search within the CSD, leading to >32 000 potential ligands. The ΔG‡ for catalysts using these ligands were calculated using B97-3c//GFN2-xTB with high success rates and good correlation compared to DLPNO-CCSD(T)/def2-TZVPP. Furthermore, machine learning models were developed based on the generated data, leading to accurate predictions of ΔG‡, with 70.6–81.5% of predictions falling within ± 4 kcal mol−1 of the calculated ΔG‡, without the need for the costly calculation of the transition state. This accuracy of machine learning models was improved to 75.4–87.8% using descriptors derived from TPSS/def2-TZVP//GFN2-xTB calculations with a minimal increase in computational time. This new workflow offers significant advantages over currently used methods due to its faster speed and lower computational cost, coupled with excellent accuracy compared to higher-level methods.
- This article is part of the themed collections: Machine Learning and Artificial Intelligence: A cross-journal collection and Catalysis Science & Technology Most Popular 2023 Articles
 Please wait while we load your content...
Please wait while we load your content...

