
An in silico predictive method to select multi-monomer combinations for peptide imprinting†
 ab
and
Boris
Mizaikoff
*ac
ab
and
Boris
Mizaikoff
*ac
Abstract
In silico methods enable optimizing artificial receptors such that constructive mimics of natural antibodies can be envisaged. The introduction of combinatorial synthesis strategies via multi-monomer combinations has improved the performance of molecularly imprinted polymers (MIP) significantly. However, it remains experimentally challenging to screen thousands of combinations resulting from a large library of monomers. The present study introduces a molecular mechanics based multi-monomer simultaneous docking approach (MMSD) to computationally screen monomer combinations according to their potential, facilitating selective molecular imprints. Thereby, the diversity of multipoint interactions realizable with a peptide surface is efficiently explored yielding how individual monomer binding capacities constructively or adversely add up when docked together. Additionally, spatially distributed molecular models were mapped for analyzing intermolecular H-bonding and hydrophobic interactions resulting from single monomer docking, as well as bi- and tri-monomer simultaneous docking. A direct impact of complex formation on the binding capacity of the resulting MIPs has been observed. In a first small-scale study, the predictive potential of the MMSD approach was validated via experimentally applied polymer combinations for peptide imprinting via the scoring functions established during the screening process. MMSD clearly enables rational design of MIPs for synthesizing more sensitive and selective artificial receptor materials especially for peptide and protein-epitope templates.
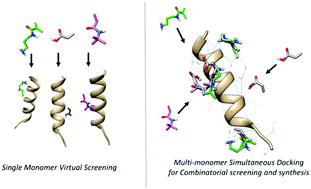
- This article is part of the themed collection: New era in advanced functional materials emerging from molecular imprinting and related techniques
 Please wait while we load your content...
Please wait while we load your content...

