
Understanding the structure–activity relationships of different double atom catalysts from density functional calculations: three general rules for efficient CO oxidation†
 *ab
*ab
Abstract
CO oxidation is considered to be a model reaction to explore the relationships between structure and catalytic performance. In this work, several double-atom catalysts (DACs) M/UiO-66 (M = Pd, Pt, Cu, Ag, Au, Zn, or Cd, UiO-66 is a classical metal–organic framework) for CO oxidation have been investigated by density functional theory (DFT) calculations. DACs were obtained by doping metal atoms into the defect sites at the zirconium oxide clusters of UiO-66. All possible mechanisms (Eley–Rideal (ER), Langmuir–Hinshelwood (LH), and termolecular ER (TER) mechanisms) for CO oxidation were considered in detail. Compared with pristine UiO-66, M/UiO-66 remarkably promoted the CO oxidation performance. Among them, the superior catalysts were Zn and Ag-DACs, whose rate-limiting energy barriers (Ebar) are as low as 0.14 and 0.22 eV, respectively. More importantly, three general rules affecting CO oxidation Ebar were proposed: (i) from the aspect of geometric structure, the corresponding Ebar is negatively correlated with the OC–O distance of the transition structure within a certain range; (ii) from the aspect of electronic structure, when the d-band center difference ratio 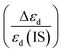 of DACs is in the range of −15% to 15%, the corresponding Ebar is less than 1.08 eV for most cases; (iii) if the Eads of individual O2 and CO molecules are similar, or the Eads of an individual O2 molecule is extremely large, the catalytic performance is expected to be satisfactory. Therefore, this work provides unique insights into the design of excellent DACs for CO oxidation, and the proposed rules can be extended to other complex reactions.
of DACs is in the range of −15% to 15%, the corresponding Ebar is less than 1.08 eV for most cases; (iii) if the Eads of individual O2 and CO molecules are similar, or the Eads of an individual O2 molecule is extremely large, the catalytic performance is expected to be satisfactory. Therefore, this work provides unique insights into the design of excellent DACs for CO oxidation, and the proposed rules can be extended to other complex reactions.

 Please wait while we load your content...
Please wait while we load your content...

