
Molecular mechanisms of amyloid formation in living systems

Abstract
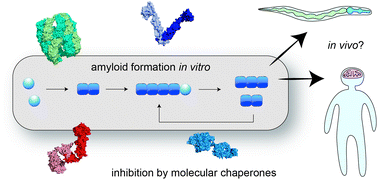
Fibrillar protein aggregation is a hallmark of a variety of human diseases. Examples include the deposition of amyloid-β and tau in Alzheimer's disease, and that of α-synuclein in Parkinson's disease. The molecular mechanisms by which soluble proteins form amyloid fibrils have been extensively studied in the test tube. These investigations have revealed the microscopic steps underlying amyloid formation, and the role of factors such as chaperones that modulate these processes. This perspective explores the question to what extent the mechanisms of amyloid formation elucidated in vitro apply to human disease. The answer is not yet clear, and may differ depending on the protein and the associated disease. Nevertheless, there are striking qualitative similarities between the aggregation behaviour of proteins in vitro and the development of the related diseases. Limited quantitative data obtained in model organisms such as Caenorhabditis elegans support the notion that aggregation mechanisms in vivo can be interpreted using the same biophysical principles established in vitro. These results may however be biased by the high overexpression levels typically used in animal models of protein aggregation diseases. Molecular chaperones have been found to suppress protein aggregation in animal models, but their mechanisms of action have not yet been quantitatively analysed. Several mechanisms are proposed by which the decline of protein quality control with organismal age, but also the intrinsic nature of the aggregation process may contribute to the kinetics of protein aggregation observed in human disease.
- This article is part of the themed collections: Amyloids and Protein Aggregation, Most popular 2022 chemical biology articles, 2022 Chemical Science HOT Article Collection and 2022 Chemical Science Perspective & Review Collection
 Please wait while we load your content...
Please wait while we load your content...

