
Computational design of next generation atom transfer radical polymerization ligands†
 *a
and
Michelle L.
Coote
*a
*a
and
Michelle L.
Coote
*a
Abstract
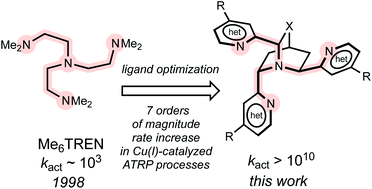
Benchmarked density functional theory is used to design and evaluate a series of novel atom transfer radical polymerization (ATRP) catalysts with a view to identifying those which best promote a model ATRP reaction between the [LCuI]+ catalyst and methyl α-bromoisobutyrate in acetonitrile. Calculations were performed at the ωB97XD/Def2TZVP//ωB97XD/SDD/6-31G(d) level of theory with CPCM solvent corrections. Locating the axial N of the tetradentate ATRP ligand at a sterically unhindered quinuclidine bridgehead site, with the cage carrying three pendant 2-pyridyl arms to serve as equatorial ligands causes a significant increase in activity. The effects are greater for the CH2-linked quinuclidine cages compared with the O-linked cages, although both systems out-perform the best available ATRP ligands in current use. Inclusion of this cage structure introduces opportunities for stereoisomerism, and while all stereoisomers examined function as effective catalysts, there is significant variation in performance. Incorporation of electron donating 4-(dialkylamino) groups on the 2-pyridyl arms improves ligand activity by stabilizing CuII over CuI, however, one needs to be careful of unfavorable steric crowding, particularly for some stereoisomers. Replacing the 2-pyridyl arms with other types of neutral heterocycles, such as NHC-containing heterocycles and imidazoles, does not appear to offer an advantage over pyridine. However, ligands with negatively charged donor nitrogens could offer 10 orders of magnitude improvement in catalyst activity over the best available neutral ligands, which could help make ATRP and other small molecule processes that employ ATRP catalysts applicable to unactivated alkyl halides.
- This article is part of the themed collection: Polymer Chemistry Most Popular 2022
 Please wait while we load your content...
Please wait while we load your content...

