
Tempering of Au nanoclusters: capturing the temperature-dependent competition among structural motifs†

Abstract
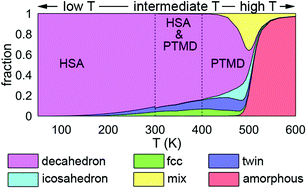
A computational approach to determine the equilibrium structures of nanoclusters in the whole temperature range from 0 K to melting is developed. Our approach relies on Parallel Tempering Molecular Dynamics (PTMD) simulations complemented by Harmonic Superposition Approximation (HSA) calculations and global optimization searches, thus combining the accuracy of global optimization and HSA in describing the low-energy part of configuration space, together with the PTMD thorough sampling of high-energy configurations. This combined methodology is shown to be instrumental towards revealing the temperature-dependent structural motifs in Au nanoclusters of sizes 90, 147, and 201 atoms. The reported phenomenology is particularly rich, displaying a size- and temperature-dependent competition between the global energy minimum and other structural motifs. In the case of Au90 and Au147, the global minimum is also the dominant structure at finite temperatures. In contrast, the Au201 cluster undergoes a solid–solid transformation at low temperature (<200 K). Results indicate that PTMD and HSA very well agree at intermediate temperatures, between 300 and 400 K. For higher temperatures, PTMD gives an accurate description of equilibrium, while HSA fails in describing the melting range. On the other hand, HSA is more efficient in catching low-temperature structural transitions. Finally, we describe the elusive structures close to the melting region which can present complex and defective geometries, that are otherwise difficult to characterize through experimental imaging.
 Please wait while we load your content...
Please wait while we load your content...

