
A DFT study on the isomerization mechanism of azobenzene derivatives on silicon substrates†

Abstract
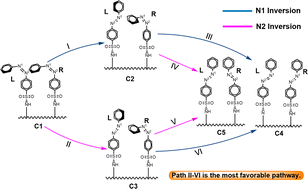
Herein, density functional theory was used to study the isomerization mechanism of azobenzenesulfonamide at the ωB97XD/6-31G(d) level. The similarities and differences among the isomerization of single- and bi-molecular azobenzenesulfonamide derivatives with silicon substrates are discussed. The calculated results show that azobenzenesulfonamide (PhN1![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N2SO2NH2Ph) has two rotation-assisted inversion isomerism modes: N1 inversion and N2 inversion. These two isomerization pathways are closely related to the substituents on the benzene ring connected to the N1 atom. The stronger the electron-withdrawing ability of the substituent, the easier it is for the N1 inversion to occur. In addition, the cis–trans isomerization mechanism of a single molecular azobenzenesulfonamide derivative with a silicon substrate is similar to that without a silicon substrate. However, the isomerization pathway of a bi-molecular azobenzenesulfonamide derivative on a silicon substrate is different from that of single-molecule isomerization. Among them, the first molecular functional group of azobenzenesulfonamide undergoes N2 inversion, the second molecular functional group then adopts the N1 inversion mode, and the free energy barriers of isomerization of the two molecular functional groups are 26.88 and 13.99 kcal mol−1, respectively. This may be related to the steric hindrance of the groups in the conversion isomers. Interestingly, compared to the isomerization of the first and second azobenzenesulfonamide molecular functional groups, the energy barrier of the second group is significantly reduced. This will provide theoretical insights into the design of azobenzene and the action of its derivatives as molecular switches in polymerization systems.
N2SO2NH2Ph) has two rotation-assisted inversion isomerism modes: N1 inversion and N2 inversion. These two isomerization pathways are closely related to the substituents on the benzene ring connected to the N1 atom. The stronger the electron-withdrawing ability of the substituent, the easier it is for the N1 inversion to occur. In addition, the cis–trans isomerization mechanism of a single molecular azobenzenesulfonamide derivative with a silicon substrate is similar to that without a silicon substrate. However, the isomerization pathway of a bi-molecular azobenzenesulfonamide derivative on a silicon substrate is different from that of single-molecule isomerization. Among them, the first molecular functional group of azobenzenesulfonamide undergoes N2 inversion, the second molecular functional group then adopts the N1 inversion mode, and the free energy barriers of isomerization of the two molecular functional groups are 26.88 and 13.99 kcal mol−1, respectively. This may be related to the steric hindrance of the groups in the conversion isomers. Interestingly, compared to the isomerization of the first and second azobenzenesulfonamide molecular functional groups, the energy barrier of the second group is significantly reduced. This will provide theoretical insights into the design of azobenzene and the action of its derivatives as molecular switches in polymerization systems.
 Please wait while we load your content...
Please wait while we load your content...

