
Modelling structural properties of cyanine dye nanotubes at coarse-grained level†
 ab
Paulo C. T.
Souza,
c
Selim
Sami,
abd
Remco W. A.
Havenith,
bde
Alex H.
de Vries
ab
and
Siewert J.
Marrink
*ab
ab
Paulo C. T.
Souza,
c
Selim
Sami,
abd
Remco W. A.
Havenith,
bde
Alex H.
de Vries
ab
and
Siewert J.
Marrink
*ab
Abstract
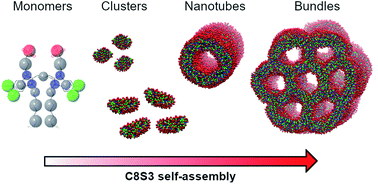
Self-assembly is a ubiquitous process spanning from biomolecular aggregates to nanomaterials. Even though the resulting aggregates can be studied through experimental techniques, the dynamic pathways of the process and the molecular details of the final structures are not necessarily easy to resolve. Consequently, rational design of self-assembling aggregates and their properties remains extremely challenging. At the same time, modelling the self-assembly with computational methods is not trivial, because its spatio-temporal scales are usually beyond the limits of all-atom based simulations. The use of coarse-grained (CG) models can alleviate this limitation, but usually suffers from the lack of optimised parameters for the molecular constituents. In this work, we describe the procedure of parametrizing a CG Martini model for a cyanine dye (C8S3) that self-assembles into hollow double-walled nanotubes. First, we optimised the model based on quantum mechanics calculations and all-atom reference simulations, in combination with available experimental data. Then, we conducted random self-assembly simulations, and the performance of our model was tested on preformed assemblies. Our simulations provide information on the time-dependent local arrangement of this cyanine dye, when aggregates are being formed. Furthermore, we provide guidelines for designing and optimising parameters for similar self-assembling nanomaterials.
- This article is part of the themed collection: Popular Advances
 Please wait while we load your content...
Please wait while we load your content...

