
Comprehensive comparison of sample preparation workflows for proteomics†
 *b
*b
Abstract
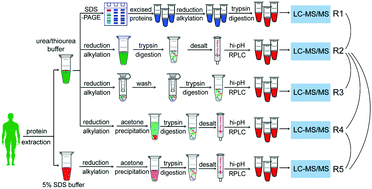
Mass spectrometry-based proteomics experiments can be subject to a large variability, which forms an obstacle to obtaining deep and accurate protein identification. Here, to obtain an optimal sample preparation workflow for in-depth proteome identification in human tissues, we systematically compared typical procedures in the four key steps during sample preparation, including two lysis buffers (5% SDS and 7M urea/2M thiourea), acetone precipitation, two proteolytic enzyme methods (in-solution digestion and FASP), and two pre-fractionation methods (SDS-PAGE and hi-pH RPLC). Compared with other methods, the procedure, including urea/thiourea as the lysis buffer, in-solution digestion, followed by hi-pH RPLC, yields an increase in proteome coverage (+15%), matched peptides (+42.4%), and significantly high protein concentrations. We also used combinations of these sample preparation methods to demonstrate a high identification rate in the range of low molecular weight (LMW), and the performance of sample preparation workflows varied between different groups of proteins. Importantly, 3 proteins defined as missing proteins (MPs) following the Human Proteome Project (HPP) guidelines were found in our data set. Overall, our findings provide an optimal sample preparation workflow for highly efficient and unbiased global proteomic analysis in human tissues.
 Please wait while we load your content...
Please wait while we load your content...