
Theoretical studies on Lennard-Jones parameters of benzene and polycyclic aromatic hydrocarbons†
 *ab
*ab
Abstract
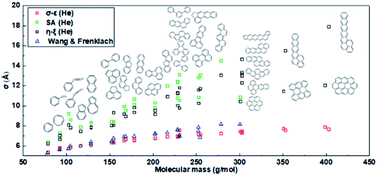
Lennard-Jones (L-J) parameters, i.e. collision diameter and well depth, of benzene and polycyclic aromatic hydrocarbons (PAHs) interacting with bath gases helium and nitrogen are studied theoretically in this work. The results of three different computing methods, called SA, σ–ε, η–ξ methods respectively, are compared with literature data. The SA method determines effective L-J parameters from the spherically averaged intermolecular potentials; the σ–ε method averages L-J parameters obtained from different relative orientations of interacting partners; and the η–ξ method uses an orientation-averaging rule on the basis of two characteristic variables η and ξ representing repulsive and attractive energy scales respectively. The σ–ε and η–ξ methods require much less computational time than the SA method due to the use of an iterative search algorithm. For validation of the L-J parameters, binary diffusion coefficients computed using L-J parameters by these three methods and those by empirical estimations are compared with experimental data from literature. Results show that while the SA method is reliable and the σ–ε method is efficient, the η–ξ method is both reliable and efficient for computing L-J parameters for benzene and PAHs, and captures the anisotropic effects of molecular structure on L-J parameters better than empirical methods.
- This article is part of the themed collection: Unimolecular reactions
 Please wait while we load your content...
Please wait while we load your content...

