
Stoichiometry modulates the optoelectronic functionality of zinc phosphide (Zn3−xP2+x)†
 a
Mischa
Flór,
a
Simon
Escobar Steinvall,
a
Maria Chiara
Spadaro,
b
Anna
Fontcuberta i Morral
ae
and
Mirjana
Dimitrievska
*a
a
Mischa
Flór,
a
Simon
Escobar Steinvall,
a
Maria Chiara
Spadaro,
b
Anna
Fontcuberta i Morral
ae
and
Mirjana
Dimitrievska
*a
Abstract
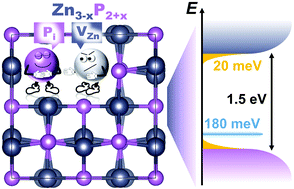
Predictive synthesis–structure–property relationships are at the core of materials design for novel applications. In this regard, correlations between the compositional stoichiometry variations and functional properties are essential for enhancing the performance of devices based on these materials. In this work, we investigate the effect of stoichiometry variations and defects on the structural and optoelectronic properties of monocrystalline zinc phosphide (Zn3P2), a promising compound for photovoltaic applications. We use experimental methods, such as electron and X-ray diffraction and Raman spectroscopy, along with density functional theory calculations, to showcase the favorable creation of P interstitial defects over Zn vacancies in P-rich and Zn-poor compositional regions. Photoluminescence and absorption measurements show that these defects create additional energy levels at about 180 meV above the valence band. Furthermore, they lead to the narrowing of the bandgap, due to the creation of band tails in the region of around 10–20 meV above the valence and below the conduction band. The ability of zinc phosphide to form off-stoichiometric compounds provides a new promising opportunity for tunable functionality that benefits applications. In that regard, this study is crucial for the further development of zinc phosphide and its application in optoelectronic and photovoltaic devices, and should pave the way for defect engineering in this kind of material.
- This article is part of the themed collection: Emerging inorganic materials in thin-film photovoltaics
 Please wait while we load your content...
Please wait while we load your content...

