
The structural pathway from its solvated molecular state to the solution crystallisation of the α- and β-polymorphic forms of para amino benzoic acid†
 ‡a
Thomas D.
Turner,
§a
Cai Y.
Ma,
a
Robert B.
Hammond,
a
Kevin J.
Roberts,
*a
Chin W.
Yong
bc
and
Ilian T.
Todorov
b
‡a
Thomas D.
Turner,
§a
Cai Y.
Ma,
a
Robert B.
Hammond,
a
Kevin J.
Roberts,
*a
Chin W.
Yong
bc
and
Ilian T.
Todorov
b
Abstract
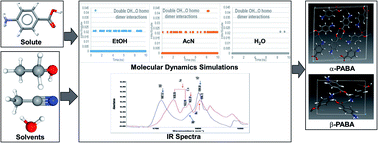
Para amino benzoic acid (PABA) has two well-characterised α- and β-polymorphic forms and, whilst both crystallise in the monoclinic space group P21/n, they have quite different crystal chemistry and crystallisability behaviour. Previous work has shown that the molecular conformation deformation energy in the crystalline state is higher for the β-form than for the α-form and that the lattice energy for the former converges more slowly than for the latter overall. This suggests that not only is there a higher barrier to crystallisation for the β-form but also that low solution supersaturations might be needed for it to preferentially nucleate. Additionally, solute cluster propensity and solute solvation energetic analysis highlight the importance of an aqueous solvation environment in inhibiting the α-form’s strong OH⋯O carboxylic acid hydrogen bond (H-bond) dimer. Despite this, the detailed molecular-scale pathway from solvated molecules to 3D crystallographic structure still remains unclear, most notably regarding how the nucleation process is activated and how, as a result, this mediates the preferential formation of either of the two polymorphic forms. Molecular dynamics (MD) simulations coupled with FTIR studies and intermolecular synthon analysis address this issue through characterisation of the propensity of the incipient bulk synthons that are important in the crystallisation of the two polymorphic forms within the solution state. MD molecular trajectory analysis within crystallisation solutions reveals a greater propensity for OH⋯O synthons (both single H-bonds and homodimers) typical of the α-form and NH⋯O synthons found in both the α- and β-forms when compared to aqueous solution but much lower propensities for the β-form’s “fingerprinting” OH⋯N and π–π stacking synthons. In contrast, data from the aqueous solution environment reveals a much greater propensity for the β-form’s π–π interaction synthons. IR dilution studies in acetonitrile in the carbonyl region reveal the presence of two C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O vibrational stretching bands, whose relative intensities vary as a function of solution dilution. These were assigned to the solvated PABA monomer and a COOH dimer of PABA. Similar data in ethanol shows a main CO stretching band with a shoulder peak suggesting a similar monomer vs. dimer speciation may exist in this solvent. The IR data is consistent with the organic solvent MD data, albeit the corresponding analysis for the aqueous solution was precluded due to the latter’s strong OH vibrational mode which restricted validation in aqueous solutions.
O vibrational stretching bands, whose relative intensities vary as a function of solution dilution. These were assigned to the solvated PABA monomer and a COOH dimer of PABA. Similar data in ethanol shows a main CO stretching band with a shoulder peak suggesting a similar monomer vs. dimer speciation may exist in this solvent. The IR data is consistent with the organic solvent MD data, albeit the corresponding analysis for the aqueous solution was precluded due to the latter’s strong OH vibrational mode which restricted validation in aqueous solutions.
- This article is part of the themed collection: Understanding Crystallisation
 Please wait while we load your content...
Please wait while we load your content...

