
Chemical bonding in actinyl(v/vi) dipyriamethyrin complexes for the actinide series from americium to californium: a computational investigation†

Abstract
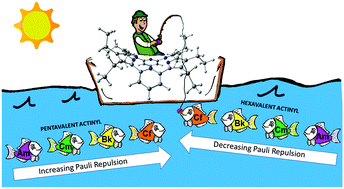
The separation of minor actinides in their dioxocation (i.e., actinyl) form in high-valence oxidation states requires efficient ligands for their complexation. In this work, we evaluate the complexation properties of actinyls including americyl, curyl, berkelyl, and californyl in their pentavalent and hexavalent oxidation states with the dipyriamethyrin ligand (L) using density functional theory calculations. The calculated bond parameters show shorter An![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Oyl bonds with covalent character and longer An–N bonds with ionic character. The bonding between the actinyl cation and the ligand anion shows a flow of charges from the ligand to actinyl in all [AnV/VIO2–L]1−/0 complexes. However, across the series, backdonation of charges from the metal to the ligand becomes prominent and stabilizes the complexes. The thermodynamic parameters in the gas phase and solution suggest that the complex formation reaction is spontaneous for [CfV/VIO2–L]1−/0 complexes and spontaneous at elevated temperatures (>298.15 K) for all other complexes. Spin–orbit corrections have a quantitative impact while the overall trend remains the same. Energy decomposition analysis (EDA) reveals that the interaction between actinyl and the ligand is mainly due to electrostatic contributions that decrease from Am to Cf along with an increase in orbital contributions due to the backdonation of charges from the actinyl metal center to the ligand that greatly stabilizes the Cf complex. The repulsive Pauli energy contribution is observed to increase in the case of [AnVO2–L]1− complexes from Am to Cf while a decrease is observed among [AnVIO2–L]0 complexes, showing minimum repulsion in [CfVIO2–L]0 complex formation. Overall, the hexavalent actinyl complexes show greater stability (increasing from Am to Cf) than their pentavalent counterparts.
Oyl bonds with covalent character and longer An–N bonds with ionic character. The bonding between the actinyl cation and the ligand anion shows a flow of charges from the ligand to actinyl in all [AnV/VIO2–L]1−/0 complexes. However, across the series, backdonation of charges from the metal to the ligand becomes prominent and stabilizes the complexes. The thermodynamic parameters in the gas phase and solution suggest that the complex formation reaction is spontaneous for [CfV/VIO2–L]1−/0 complexes and spontaneous at elevated temperatures (>298.15 K) for all other complexes. Spin–orbit corrections have a quantitative impact while the overall trend remains the same. Energy decomposition analysis (EDA) reveals that the interaction between actinyl and the ligand is mainly due to electrostatic contributions that decrease from Am to Cf along with an increase in orbital contributions due to the backdonation of charges from the actinyl metal center to the ligand that greatly stabilizes the Cf complex. The repulsive Pauli energy contribution is observed to increase in the case of [AnVO2–L]1− complexes from Am to Cf while a decrease is observed among [AnVIO2–L]0 complexes, showing minimum repulsion in [CfVIO2–L]0 complex formation. Overall, the hexavalent actinyl complexes show greater stability (increasing from Am to Cf) than their pentavalent counterparts.
 Please wait while we load your content...
Please wait while we load your content...

