
Ranking the synthesizability of hypothetical zeolites with the sorting hat†

Abstract
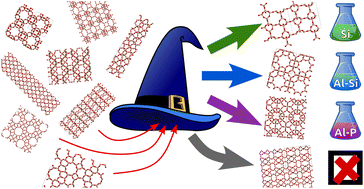
Zeolites are nanoporous alumino-silicate frameworks widely used as catalysts and adsorbents. Even though millions of siliceous networks can be generated by computer-aided searches, no new hypothetical framework has yet been synthesized. The needle-in-a-haystack problem of finding promising candidates among large databases of predicted structures has intrigued materials scientists for decades; yet, most work to date on the zeolite problem has been limited to intuitive structural descriptors. Here, we tackle this problem through a rigorous data science scheme—the “Zeolite Sorting Hat”—that exploits interatomic correlations to discriminate between real and hypothetical zeolites and to partition real zeolites into compositional classes that guide synthetic strategies for a given hypothetical framework. We find that, regardless of the structural descriptor used by the Zeolite Sorting Hat, there remain hypothetical frameworks that are incorrectly classified as real ones, suggesting that they might be good candidates for synthesis. We seek to minimize the number of such misclassified frameworks by using as complete a structural descriptor as possible, thus focusing on truly viable synthetic targets, while discovering structural features that distinguish real and hypothetical frameworks as an output of the Zeolite Sorting Hat. Further ranking of the candidates can be achieved based on thermodynamic stability and/or their suitability for the desired applications. Based on this workflow, we propose three hypothetical frameworks differing in their molar volume range as the top targets for synthesis, each with a composition suggested by the Zeolite Sorting Hat. Finally, we analyze the behavior of the Zeolite Sorting Hat with a hierarchy of structural descriptors including intuitive descriptors reported in previous studies, finding that intuitive descriptors produce significantly more misclassified hypothetical frameworks, and that more rigorous interatomic correlations point to second-neighbor Si–O distances around 3.2–3.4 Å as the key discriminatory factor.
 Please wait while we load your content...
Please wait while we load your content...

