
Limitations of machine learning models when predicting compounds with completely new chemistries: possible improvements applied to the discovery of new non-fullerene acceptors†
 ‡*a
and
Alessandro
Troisi
a
‡*a
and
Alessandro
Troisi
a
Abstract
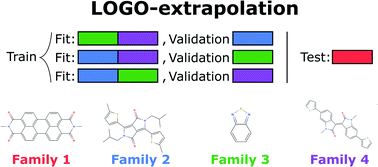
We try to determine if machine learning (ML) methods, applied to the discovery of new materials on the basis of existing data sets, have the power to predict completely new classes of compounds (extrapolating) or perform well only when interpolating between known materials. We introduce the leave-one-group-out cross-validation, in which the ML model is trained to explicitly perform extrapolations of unseen chemical families. This approach can be used across materials science and chemistry problems to improve the added value of ML predictions, instead of using extrapolative ML models that were trained with a regular cross-validation. We consider as a case study the problem of the discovery of non-fullerene acceptors because novel classes of acceptors are naturally classified into distinct chemical families. We show that conventional ML methods are not useful in practice when attempting to predict the efficiency of a completely novel class of materials. The approach proposed in this work increases the accuracy of the predictions to enable at least the categorization of materials with a performance above and below the median value.
- This article is part of the themed collection: Celebrating our 2024 Prizewinners
 Please wait while we load your content...
Please wait while we load your content...

