
The soluble N-terminal autoinhibitory module of the A1 domain in von Willebrand factor partially suppresses its catch bond with glycoprotein Ibα in a sandwich complex
 c
and
Lining Arnold
Ju
*abdef
c
and
Lining Arnold
Ju
*abdef
Abstract
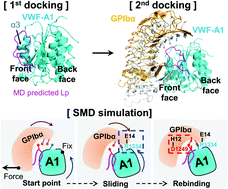
von Willebrand factor (VWF) senses and responds to the hemodynamic forces to interact with the circulatory system and platelets in hemostasis and thrombosis. The dark side of this mechanobiology is implicated in atherothrombosis, stroke, and, more recently, the COVID-19 thrombotic symptoms. The force-responsive element controlling VWF activation predominantly resides in the N terminal auto-inhibitory module (N-AIM) flanking its A1 domain. Nevertheless, the detailed mechano-chemistry of soluble VWF N-AIM is poorly understood at the sub-molecular level as it is assumed to be unstructured loops. Using the free molecular dynamics (MD) simulations, we first predicted a hairpin-like structure of the soluble A1 N-AIM derived polypeptide (Lp; sequences Q1238–E1260). Then we combined molecular docking and steered molecular dynamics (SMD) simulations to examine how Lp regulates the A1–GPIbα interaction under tensile forces. Our simulation results indicate that Lp suppresses the catch bond in a sandwich complex of A1–Lp–GPIbα yet contributes an additional catch-bond residue D1249. To experimentally benchmark the binding kinetics for A1–GPIbα in the absence or presence of Lp, we conducted the force spectroscopy—biomembrane force probe (BFP) assays. We found similar suppression on the A1–GPIbα catch bond with soluble Lp in presence. Clinically, as more and more therapeutic candidates targeting the A1–GPIbα axis have entered clinical trials to treat patients with TTP and acute coronary syndrome, our work represents an endeavor further towards an effective anti-thrombotic approach without severe bleeding side effects as most existing drugs suffer.
- This article is part of the themed collection: 2022 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

